The mission of our research program is to understand the molecular basis of human diseases via state-of-the-art molecular biophysics and computational biology/biomechanics approaches.
Mechanical phenomena affect nearly every aspect of cellular biology and function, yet the underlying mechanisms of how mechanical forces and biochemical signals interact is not clearly understood. By understanding the regulatory mechanisms involved in these cellular signaling processes, we aim to shed light on their role in human disease. Currently, our lab is engaged in the following specific projects:
I. Molecular pathways involved in cell mechano-chemical signal transduction (or “mechanotransduction”), coupling the extracellular matrix to the nucleus
II. Bacterial mechanotransduction and microbiomes
III. Deep learning for biology (“deep proteomics” and “deep genomics”) and medicine (“deep medicine”)
IV. Multiscale models of cardiovascular biomechanics
These projects are briefly described below:
I. LOOKING “UNDER THE HOOD” OF CELLULAR MECHANOTRANSDUCTION
Mechanics of Integrin-Mediated Focal Adhesions. The underlying mechanics and mechanisms of mechanotransduction are not yet clearly understood. One hypothesis is that forces transmitted via individual proteins, either at the site of cell adhesion to its surroundings or within the stress-bearing members of the cytoskeleton, cause conformational changes that change the binding affinity of these proteins to other intracellular molecules. This altered equilibrium state can subsequently initiate biochemical signaling cascades or produce immediate structural changes. Force-induced conformational changes (‘deformations’) in proteins may play a critical role in initiating and controlling cell signaling pathways. To understand mechanotransduction at the molecular level requires detailed analysis of protein molecular conformational changes that occur in response to forces, which can be exerted by extracellular matrix through the cellular membrane or the cytoskeleton.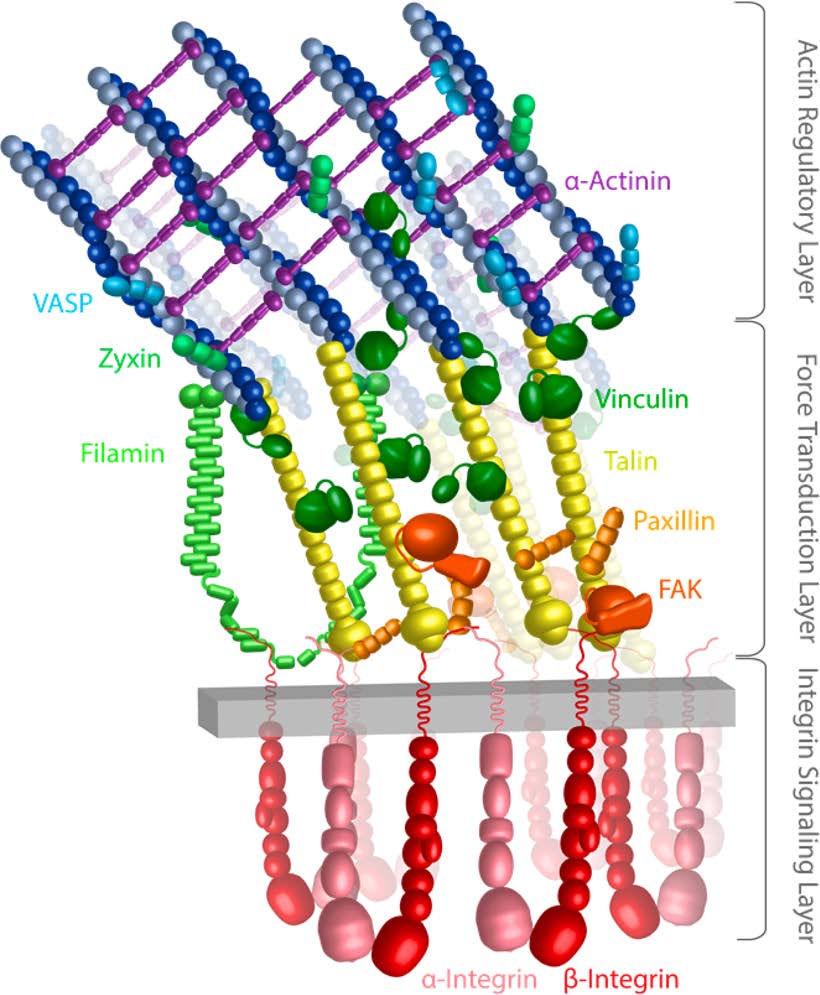
Cytoskeletal Organization and Mechanics. We study actin cross-linking by α-actinin and its further reinforcement with vinculin (Shams et al. Submitted).
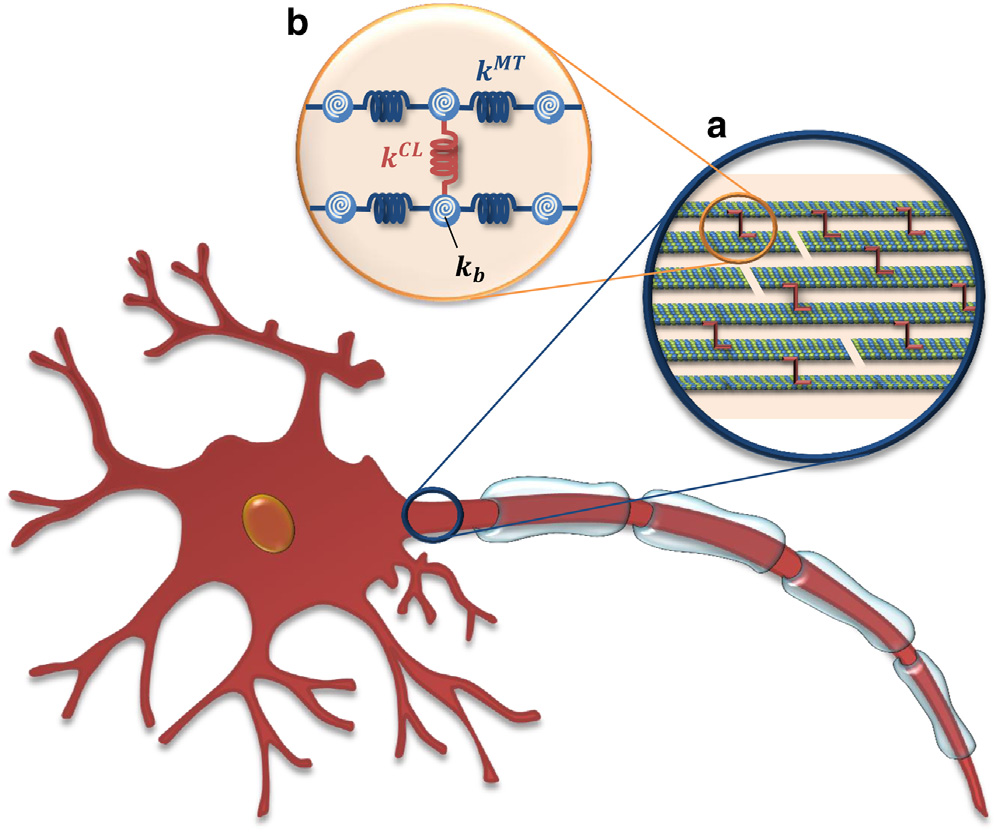
Axonal Microtubule Mechanics. We also investigate the mechanical properties of microtubules and microtubular bundles, e.g. in neuronal axons under tension (Mehrbod et al. 2011; Peter et al. 2012) along with buckling (Soheilypour et al. 2015) and torsional behavior (Lazarus et al. 2015). Axonal microtubules (MTs) are bundled by interconnections mediated by MT-associated protein (MAP) tau and form polarized arrays located in the interior portion of the axon. While today’s imaging methods are not able to detect these microscopic damages to axons, computational modeling can efficiently simulate the neuronal components subjected to external forces and substantially improve contemporary understanding of the underlying damage mechanisms. We have developed a computational model of axonal MTs to explore the biomechanical behavior of axon components under excessive compressive, tensile, and torsional forces. This work has implications in our understanding of traumatic brain injury.
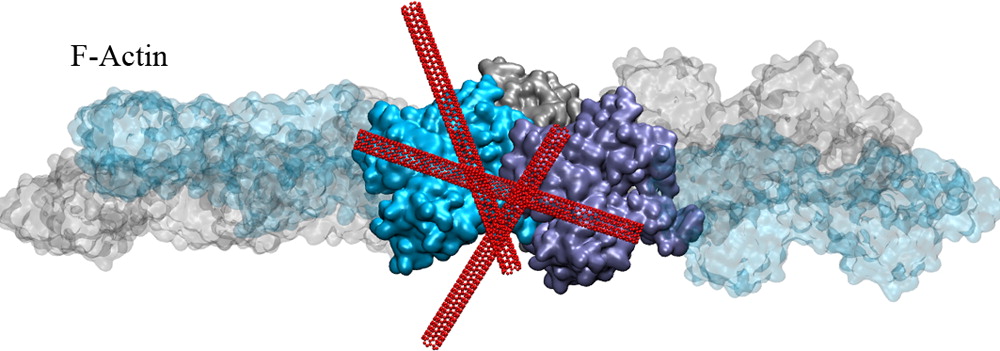
Actin Reorganization through Dynamic Interactions with Single-Wall Carbon Nanotubes.In another project, we investigated the direct interaction of actin with single-walled carbon nanotubes (SWCNT) using all-atom molecular dynamics simulations. Single-wall carbon nanotubes (SWCNTs) have been widely used for biological applications in recent years, and thus, it is critical to understand how these inert nanomaterials influence cell behavior. We showed that actin can stably bind to the SWCNT surfaces via hydrophobic interactions but still allows nanotubes to slide and rotate on the actin surface. Our results establish several nanoscale conformational changes for the actin–SWCNT complexes, and we suggest these changes likely induce reorganization of actin filaments observed at larger scales (Shams et al. 2013).
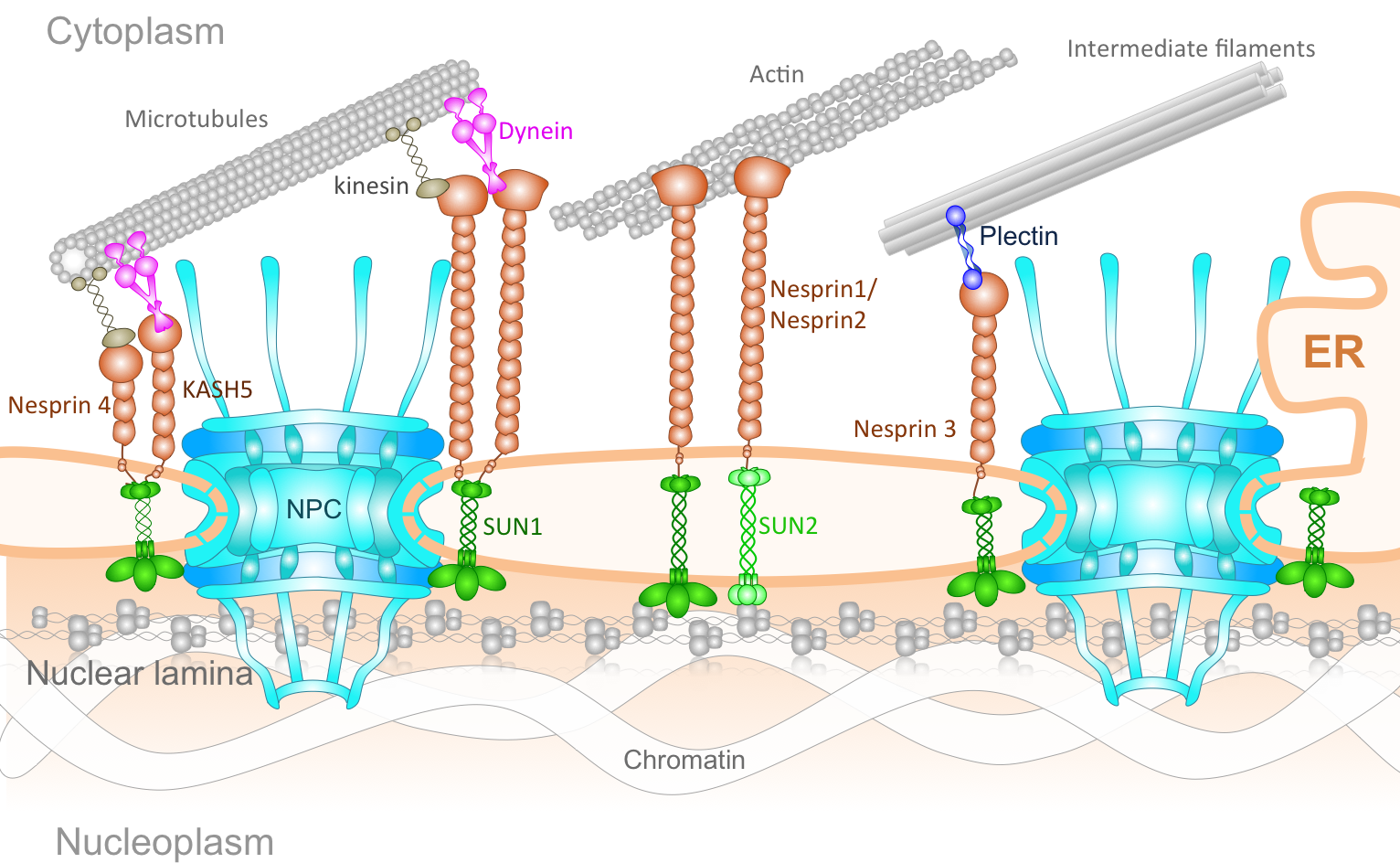
The Connection of Cytoskeleton to Nucleus. The genetic information of eukaryotic cells is enclosed within a double-layered nuclear envelope, which comprises an inner and outer nuclear membrane. Several transmembrane proteins locate to the nuclear envelope; however, only two integral protein complexes span the nuclear envelope and connect the inside of the nucleus to the cytoplasm. The nuclear pore complex (NPC) acts as a gateway for molecular exchange between the interior of the nucleus and the cytoplasm, whereas so-called LINC complexes physically link the nucleoskeleton and the cytoskeleton. The assembly of NPCs and their even distribution throughout the nuclear envelope is dependent on components of the LINC complex. Additionally, LINC complex formation is dependent on the successful localization of inner nuclear membrane components of LINC complexes and their transport through the NPC. Furthermore, the architecture of the nuclear envelope depends on both protein complexes. Finally, the LINC complexes can affect nucleocytoplasmic transport through the NPC. Understanding the NPC and LINC complexes, the relation between them and how they govern the mechanics and transport of nucleo-cytoskeletal connection offers important insight into mechanobiology of several human diseases.
Mechanochemistry of the Nuclear Pore Complex. Nuclear pore complex (NPC) is the sole gateway for bidirectional transport of vital cargos, ranging from different functional proteins to RNAs and ribosomes, between the cytoplasm and the nucleus in eukaryotic cells. The complex, yet delicate, geometry of the NPC and the fine spatiotemporal resolution at which the nucleocytoplasmic transport takes place have so far hindered the direct, experimental investigation of this exquisite nanopore. Using a hybrid of state-of-the-art computational modeling approaches, ranging from finite element (Wolf et al. 2008) to coarse-grained Brownian dynamics (Moussavi-Baygi et al. 2016) to molecular dynamics techniques (Zhao et al. 2014) to new agent-based modeling methods (Azimi et al. 2014), we study the structure and function of the nuclear pore complex and the dynamics of nucleocytoplasmic traffic. Understanding the biomechanics of the nuclear pore complex and nucleocytoplasmic transport will broadly impact our understanding of viral diseases and will revolutionize therapeutic approaches (e.g. gene therapy) and open the door to many industrial applications of biomimetic artificial nanopores.
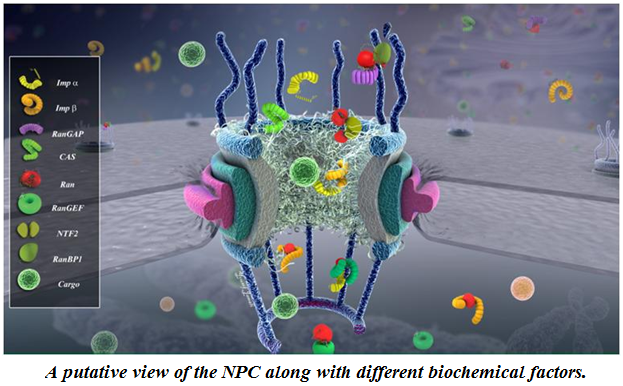
mRNA export and quality control. Following transcription, messenger ribonucleic acids (mRNAs) are transported to the cytoplasm to transfer genetic information and direct synthesis of functional proteins. Export of mRNAs into the cytoplasm is meticulously quality controlled by intricate mechanisms in eukaryotic cells. Despite the significance of these processes, many aspects of them are still poorly understood. We are developing and employing different computational techniques, such as agent-based modeling and molecular dynamics, to shed light on these essential processes in mRNA biogenesis (Soheilypour et al. 2016).

II. BACTERIAL MECHANOTRANSDUCTION AND MICROBIOMES
Bacterial cells involve mechanisms that are similar to mammalian cells to some extent but are also different in many aspects. Bacterial adhesion, which is subject to biochemical and mechanical factors, is relevant in the pathogenesis of bacterial infections and community-level dynamics such as the human microbiome. Bacteria are lined with cell wall anchored proteins, which play a critical role in the molecular basis of adhesion to many surfaces and proteins. A broad range of human diseases is associated with bacterial infections, often initiated by specific adhesion of a bacterium to the target environment. Despite the significant role of bacterial adhesion in human infectious diseases, details and mechanisms of bacterial adhesion have remained elusive. We combine experimental and computational methods to study the adhesion characteristics of Staphylococcus aureus.
Staphylococcus Aureus Adhesion at the Cellular and Molecular Levels.We are exploring the interaction of S. Aureus bacterial cells with nanostructures of various size and geometry (Jahed et al. 2014), and studying force sensing of S. Aureus surface proteins by examining the interplay of fibronectin binding protein A (FnBPA) of S. Aureus with fibrinogen, fibronectin and integrin. We are also examining the structural mechanics of a characteristic bacterial collagen-binding adhesion (CNA) of Staphylococcus Aureus (Madani et al. 2017).
III. DEEP LEARNING FOR BIOLOGY AND MEDICINE
“Deep Genomics” and “Deep Proteomics”: Application of Deep Learning and Language Processing in Biology.
A broad and simple definition of `language’ is a set of sequences constructed from a finite set of symbols. By this definition, biological sequences, natural languages, and many sequential phenomena exist in the world can be considered as languages. Although this definition is simple, it includes languages employing very complicated grammars in the creation of their sequences of symbols. Examples are biophysical principles governing biological sequences (e.g., DNA, RNA, and protein sequences), as well as grammars of natural languages determining the structure of clauses and sentences (Asgari and Mofrad, 2019).
We have a data-driven and language-agnostic point of view in the processing of biological sequences.
We use two main strategies for this purpose:
- (i) character-level or more accurately subsequence-level processing
of languages, which allows for simple modeling of the sequence similarities based on local information or bag-of-subsequences - (ii) language model-based representation of sequences encoding contextual information of sequence elements trained using neural networks.
We work on data-driven and subsequence-based language processing to address important research problems in proteomics and metagenomics:
(i) Machine Learning and Deep Language Processing for Protein Informatics
One of the main challenges in proteomics is that there exists a large gap between the number of known protein sequences and known protein tertiary structures and functions. The central question we address is how to efficiently use a large amount of existing primary sequences to achieve a better performance in the structural and functional annotation of protein sequences. We proposed a data-driven subsequence-based representations of protein sequences and their language model-based embeddings trained over a large dataset of protein sequences, called protein vectors (Asgari and Mofrad, 2015). In addition, we introduced a motif discovery approach benefiting from probabilistic segmentation of protein sequences to find functional and structural motifs (Asgari et al., 2019). This segmentation is also inferred from large protein sequence datasets in a data-driven manner. Protein vectors had a seminal contribution in protein informatics and now are widely used for machine learning based protein structure and function annotations.
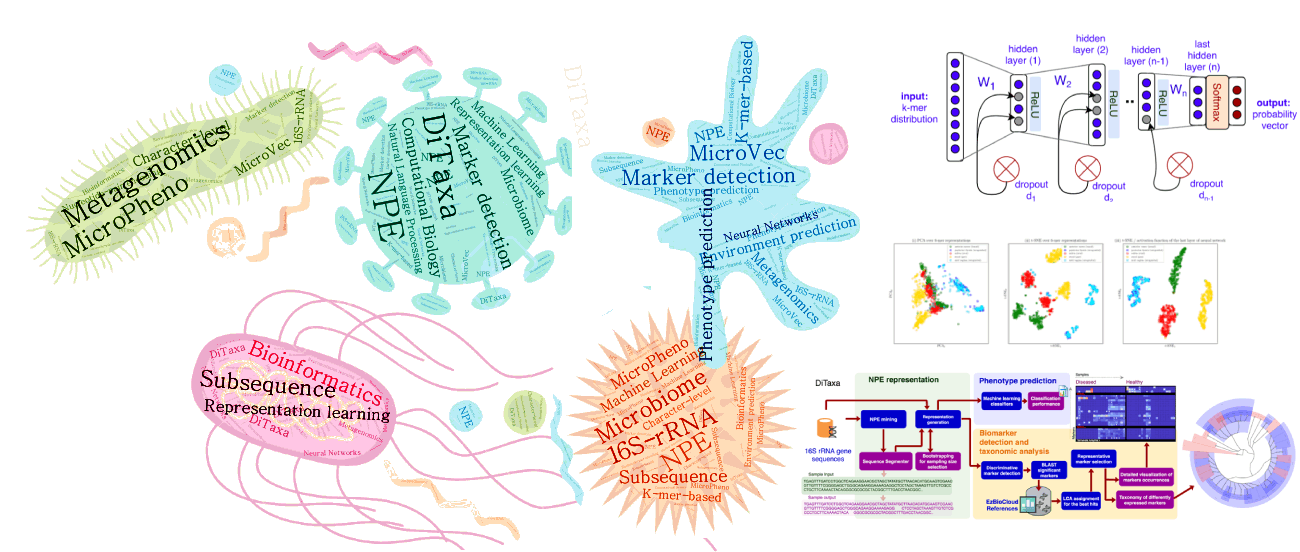
(ii) Machine Learning and Deep Language Processing for Microbial Informatics
Microbial communities exist almost on every accessible surface on earth, supporting, regulating, and even causing unwanted conditions (e.g. diseases) to their hosts and environments. Detection of the host phenotype and the phenotype-specific taxa from the microbial samples is one of the prominent problems in metagenomics. For instance, identifying distinctive taxa for microbiome-related diseases is considered key to the establishment of diagnosis and therapy options in precision medicine and imposes high demands on the accuracy of microbiome analysis techniques. We develop data-driven methods to perform machine learning on 16S rRNA sequencing, which is the most cost-effective approach for sequencing of microbial communities to date.
We have propose alignment- and reference- free methods, called MicroPheno (Asgari et al., 2018) and DiTaxa (Asgari et al., 2018), which are mainly designed for microbial phenotype and biomarker detection, respectively.
MicroPheno is a k-mer based approach achieving the state-of-the-art performance in the host phenotype prediction from 16S rRNA outperforming conventional features, Operational Taxonomic Units (OTUs) (Asgari et al., 2018).
DiTaxa, substitutes standard OTU-clustering by segmenting 16S rRNA reads into the most frequent variable-length subsequences. We compared the performance of DiTaxa to the state-of-the-art methods in phenotype and biomarker detection, using human-associated 16S rRNA samples for periodontal disease, rheumatoid arthritis, and inflammatory bowel diseases, as well as a synthetic benchmark dataset. DiTaxa performed competitively to MicroPheno (the state-of-the-art approach) in phenotype prediction while outperforming the OTU-based state-of-the-art approach in finding biomarkers in both resolution and coverage evaluated over known links from literature and synthetic benchmark datasets (Asgari et al., 2018).
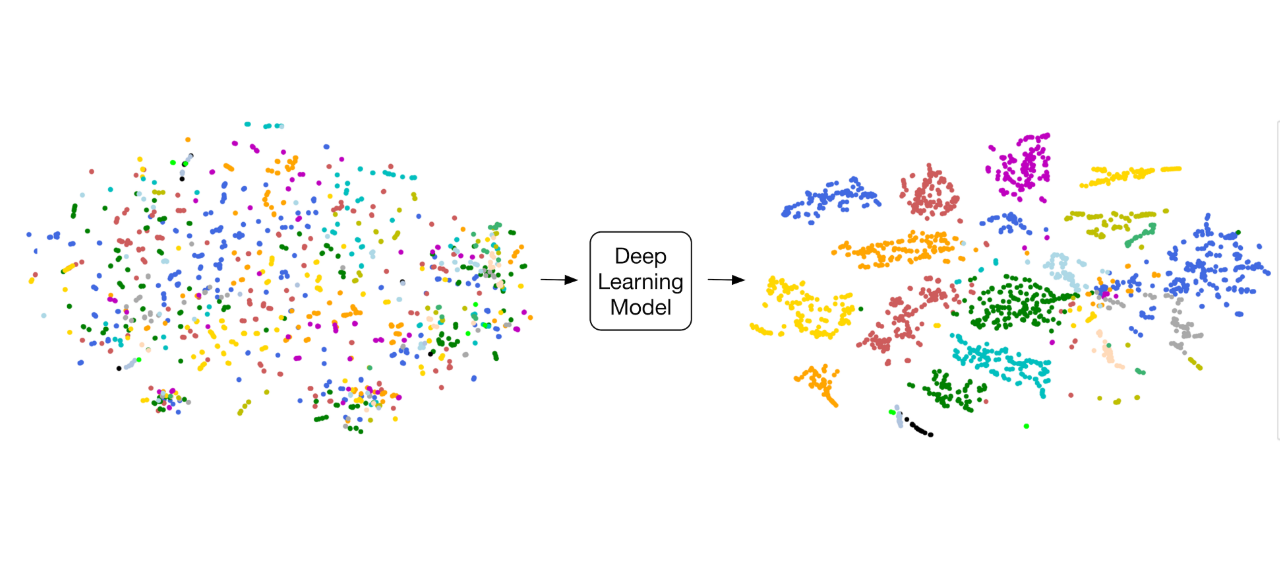
“Deep Medicine”: Deep Learning for Clinical Classifications and Diagnostics. Advances in artificial intelligence have the potential for transformative change in the field of medicine. We are using computer vision techniques for varying imaging modalities from CT, MR, ultrasound, and X-ray to automate disease classification and segmentation (Madani et al. Deep Echocardiography. 2018).
In addition, through the use of unsupervised learning, we are gaining further insights in analyzing cardiovascular disease. Many of our deep learning models outperform clinician performance for routine tasks. However, we hope to focus our computational research efforts to assist and expand the capabilities of modern medicine to improve patient care.
IV. MECHANICS OF HUMAN DISEASE
A long-term goal of our research program has been to understand the role of mechanics and mechanotransduction in human diseases, in particular, cardiovascular diseases like atherosclerosis and aortic valve calcification, and cancer.
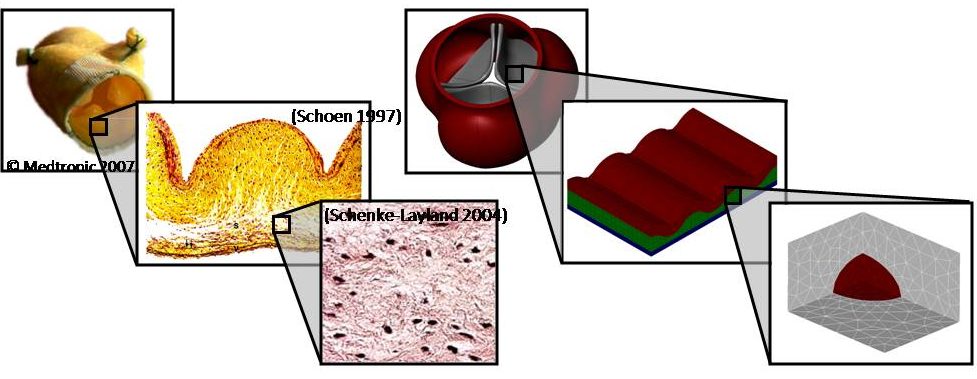
Multiscale Mechanics of the Aortic Heart Valve: A Mechanotransduction Perspective on Calcific Aortic Stenosis. The human heart is a pump system consisting of four chambers and four valves. When functioning correctly, the valves open widely to allow blood through and seal securely shut. Valvular disease inhibits a valve’s ability to open and close, decreasing the efficiency of the heart and leading to a variety of other cardiovascular disorders. Aortic valves (AV) control the flow of oxygen-rich blood from the left ventricle to the aorta and thereby the rest of the body. Normally, the aortic valve functions very efficiently, providing negligible resistance to forward flow and allowing minimal backflow. The mechanical function of the aortic valve can, however, be affected by pathological conditions. The most common valvular disease is calcific aortic stenosis (CAS). In CAS, the aortic valve undergoes changes very similar to those seen in atherosclerosis. First, inflammatory cells migrate to the site. Monocytes adhere to the endothelial layer, infiltrate it, and differentiate into macrophages. The macrophages send intracellular signals to nearby fibroblasts, causing the fibroblasts to promote cellular proliferation and matrix remodeling. Macrophages add calcium deposits to the matrix. Eventually, the remodeled matrix and calcium deposits build up to yield a thickened, stiffened leaflet. These changes can affect mechanical function, a result known as stenosis. To understand the mechanisms of calcific aortic stenosis, and to evaluate methods of prevention and treatment for this disease, we develop computational models of aortic valve mechanics.
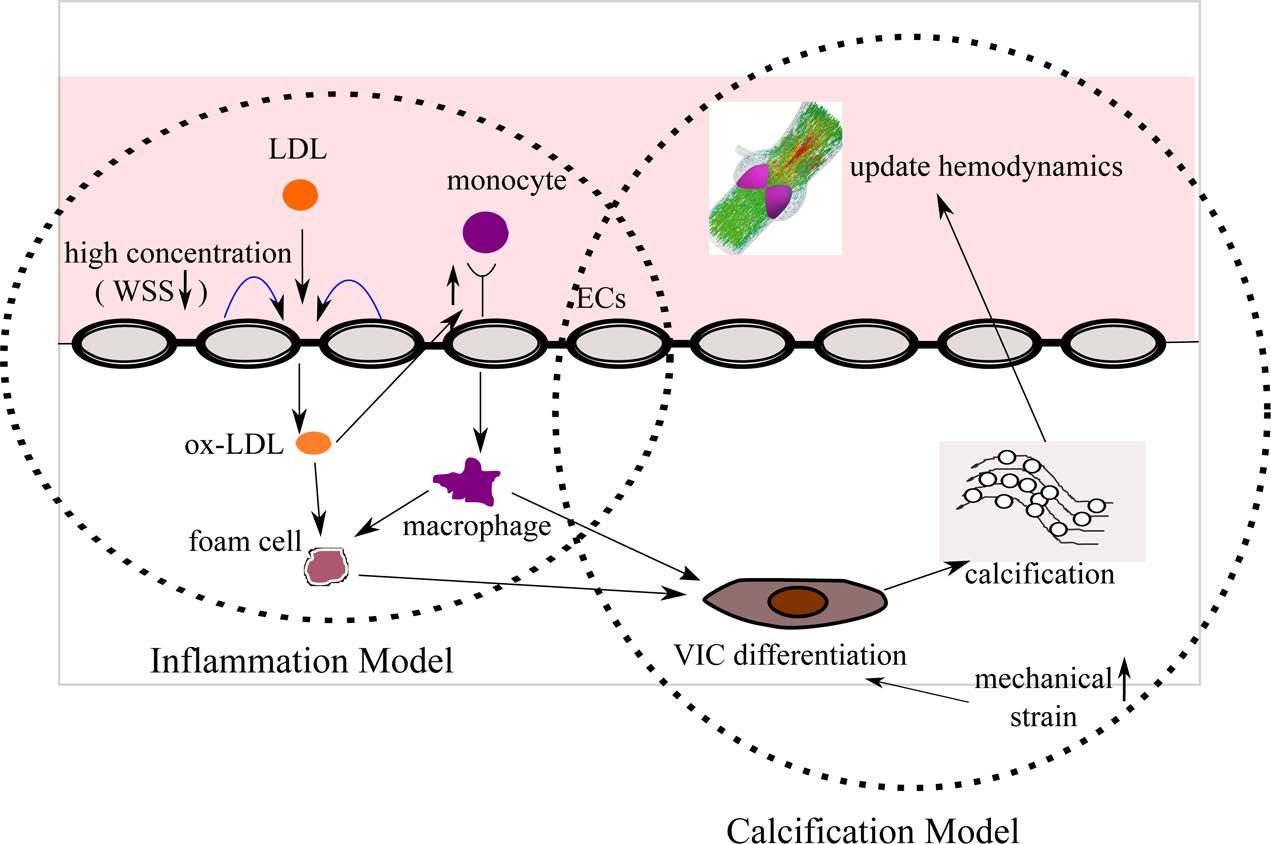
Role of Mechanics and Mechanotransduction in Arterial Disease. We have a long-term interest in understanding and characterization of atherosclerotic plaques. Due to focal nature of this disease, mechanical factors (namely, arterial wall mechanics, hemodynamics, and mass transport patterns) are widely believed to play a key role in initiation and progression of atherosclerosis. Rigorous assessment of potential links between such mechanical factors and atherosclerosis, however, requires detailed studies using realistic, subject-specific models. Over the past several years, we have developed models for computational simulation of hemodynamics and wall mechanics in patient-specific carotid bifurcations.
Last updated October 2018